HomeGesundheitCorona-VirusWie immer gefährlicher, übertragbarer, tödlicher: C.1.2: Begrüßen Sie mit uns die SARS-CoV-2 Variante Nummer 1.304!!!
August 31, 2021
Wie immer gefährlicher, übertragbarer, tödlicher: C.1.2: Begrüßen Sie mit uns die SARS-CoV-2 Variante Nummer 1.304!!!
Heureka.
Es gibt eine neue.
Alpha, Beta, Delta, sie sind Geschichte. Lambda, einst der Shooting-Star am Varianten-Himmel, von dem sich die Mortalitätsenthusiasten und Intensivbettenfanatiker so viel erwartet haben, weigert sich standhaft, im Wesentlichen Peru in relevanter Menge zu verlassen und nach Europa zu reisen.
Aber jetzt gibt es C.1.2!
C.1.2
Die MS-Medien-Euphorie ist ausgebrochen:
Laut einer Studie könnte C.1.2 noch ansteckender sein als die Delta Variante (Web.de);
In den vergangenen Monaten hat sich die Variante C.1.2 weiter ausgebreitet, vor allem in Südafrika (t-online);
C.1.2-Impfstoff könnte bei neuer Mutanten nicht wirken (ruhr24);
Neue Corona-Variante C.1.2 beschäftigt Wissenschaftler in Südafrika – angesichts der hohen Mutationsrate haben Wissenschaftler … ein besonderes Auge auf … C.1.2 (Stern)
C.1.2 – Neue Corona-Variante hat hohes Mutationspotential (n-TV);
C.1.2 – gefährlicher als Delta? Stark mutierte Corona-Variante entdeckt (nochmal n.TV);
C.1.2 aus Südafrika: Corona-Variante mit ungewöhnlich vielen Mutationen aufgetaucht (Schweriner Volkszeitung);
In einer alarmierenden Studie berichten Wissenschaftler aus Südafrika über eine neue Corona-Variante (Radio Hamburg);
Nach Delta: Forscher identifizieren neue ‘besorgniserregende’ Corona-Variante C.1.2 (Nürnberger Nachrichten):
Der Fundus von C.1.2 besteht derzeit aus 70! Sequenzen, davon sind 15 Sequenzen als C.1.2 ausgewiesen, 55 sind C.1.2 nur zugeordnet. Ein wichtiger Unterschied, zu dem wir noch kommen. C.1.2 ist eine Bezeichnung, die der Nomenklatura der sogenannte Pango Lineage entstammt, einem Versuch, das Mutationschaos, das sich bei RNA-Viren und DNA-Viren einstellt, mehr oder minder in den Griff zu bekommen. Die Zuordnung einer Sequenz zu einer Lineage ist klar geregelt. Es gibt ein Set von Regeln, das über 10 Seiten ausgebreitet wird, und das hier nachgelesen werden kann. Notwendig ist diese Nomenklatur, weil das Genom von SARS-CoV-2 rund 30.000 Nukleotide umfasst und somit rund 30.000 potentielle Orte, an denen sich eine Mutation einstellen kann. Nicht nur können sich an 30.000 verschiedenen Stellen im Genom von SARS-CoV-2 Mutationen einstellen, es können sich auch in unterschiedlichen Sequenzen an unterschiedlichen Stellen unterschiedlich viele Mutationen einstellen. Deshalb ist es notwendig, eine Methode zu finden, um die vielen möglichen Mutationen, die sich auf einem Genom oder auf einer Genomsequenz einfinden können, zuzuordnen oder genau auszuweisen.
Pango Lineage ist ein solches System. C.1.2 ist eine Benennung, aus dem System der Pango Lineages. Sie weist C.1.2 als zweiten Abkömmling des ersten Abkömmlings von C. C.1 ist identisch mit B.1.1.1, dem ersten Abkömmling des ersten Abkömmlings des ersten Abkömmlings von B. Drei Ziffern nach dem Buchstaben ist in der Nomenklatur jeweils Schluss, und es wird im nächsten Buchstaben neu begonnen. B.1.1.7, Alpha für die WHO und “die britische Mutante”, für Hirnis in deutschen Redaktionsstuben, ist der siebte Abkömmling des ersten Abkömmlings von A.1.1.1; Schon diese Nomenklatur macht deutlich, dass sich die Gensequenzen, die hier geordnet werden, in einem Punkt unterscheiden, während sie in anderen identisch sind, denn um einen Abkömmling von A.1.1.1 unterscheiden zu können, muss es eine Anzahl von Gemeinsamkeiten und eine Reihe von Unterschieden geben. Auf einem Genom mit 30.000 Mutationsmöglichkeiten, kein Problem. Das, was in Medien als Delta oder Alpha oder nun als C.1.2 bezeichnet wird, basiert in der Regel auf Gensequenzen, die bestimmte, C.1.2 definierende Mutationen teilen, während sie in anderen Teilen ihres Genoms durchaus verschieden sein können.
Die folgende Abbildung zeigt das System der Zuordnung, das in Pango Lineages zum Einsatz kommt, eine Mischung aus Entwicklungsdynamik und Hierarchie:
Die Ausweisung einer neuen Lineage und die Bestimmung der sie definierenden Merkmale ist ein Prozess, der von einer dazu eingesetzten Arbeitsgruppe durchgeführt wird. Nachdem eine Lineage ausgewiesen ist, können Zuordnungen erfolgen, Voraussetzung ist, dass die Gensequenz mindestens 95% des Genoms von SARS-CoV-2 umfasst. Gensequenzen, die nur Teile des Genoms umfassen, werden über die Software “Pangolin” zugewiesen. Die Software basiert darauf, die Lineage für die Mutationen, die in einer Gensequenz gefunden werden, zu finden, mit denen die Gensequenz die meisten Übereinstimmung aufweist.
Bislang gibt es 1.304 Pango Lineages, also Varianten von SARS-CoV-2. C.1.2 ist eine davon und nicht einmal eine bemerkenswerte. Der von Aine O’Toole, Oliver G. Pybus, Michael E. Abram, Elizabeth J. Kelly und Andrew Rambaut verfasste Beitrag “Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences” ist ein guter Einstieg, um ein Gefühl dafür zu bekommen, was sich eigentlich hinter der Bezeichnung “B.1.1.7” im Besonderen bzw. einer Pango lineage im Allgemeinen bzw. hinter der sehr kruden Bezeichnung “Alpha” oder “Delta”, die die WHO bevorzugt, versteckt.
In diesem Beitrag findet sich die folgende Abbildung:
Dargestellt sind hier Häufigkeiten für Mutationen auf bestimmten Positionen im Genom von Alpha, Beta, Gamma und Delta. Mutationen können als synonyme oder nicht-synonyme Mutation auftreten oder als Einfügung bzw. Löschung einer Position im Genom. Bei synonymen Mutationen wird in der Regel lediglich ein Basenpaar im dritten Nukleotid eines Kodons verändert. Aminosäuren in RNA bestehen aus mehreren Kodons. Eine synonyme Mutation ändert nichts an der Aminosäure. Eine nicht synonyme Mutation verändert die Aminosäure, in dem entweder ein Nukleotid gelöscht oder ergänzt wird. Nicht synonyme Mutationen sind deutlich seltener als synonyme Mutationen und im Gegensatz zu synonymen Mutationen relevant.
O’Toole und Kollegen (2021) finden z.B. für das Spike-Protein in 89,2% der Positionen eine Mutation (1138 von 1274)., wenn sie die 345.356 SARS-CoV-2 Genome, die in GISAID gespeichert sind, analysieren. Unter 458.622 Gensequenzen von SARS-CoV-2, die einer Lineage im Pango System zugeordnet sind, finden die Autoren 31.940 Nukleotid-Sequenzen, die dem Spike-Protein zugeordnet sind, davon finden sich 2.600 in mehr als einer Lineage, vier finden sich in mehr als 50 Lineages und eine hat es in 658 Lineages geschafft. Das zeigt sehr deutlich, dass eine Sequenz, die b.1.1.7 zugeordnet wird, nicht grundverschieden von einer Sequenz ist, die C.1.2 zugeordnet wird. Die “neuen” Varianten sind nur insofern neue Varianten als sie den Teilen von bislang vorhandenen Varianten, die sie nach wie vor umfassen, neue Mutationen hinzugesellen. Die Pango Nomenklatur trägt dem Rechnung. C.1.2 ist also keine Mutation, die vom Himmel gefallen ist, sie ist Teil einer Evolution, die mit A.1 begonnen hat. C.1.2 enthält weiterhin Teile von A.1 und von B.1 und von B.1.1.1 und zusätzlich ein paar neue Mutationen, die ausreichen, um C.1.2 als ausreichend verschieden von B.1.1.1 anzusehen und eine neue Lineage, Lineage Nr. 1.304 zu eröffenen: C.1.2.
Für diese neue Lineage gibt es derzeit 70 Sequenzen. Das hindert die Hirnis, die in Redaktionen sitzen, aber nicht daran, schon einmal den Panikknopf umzustellen und ihr übliches Lamento anzustimmen, das, das sie immer anstimmen: gefährlicher, übertragbarer, tödlicher. Alle Beiträge haben wiederum die gerade von Catherine Scheepers und … bis Jinal N. Bhiman (2021) veröffentlichte Arbeit mit dem Titel “The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection” zum Gegenstand, in der die Autoren von ihren Analysen berichten, die sie auf Basis von 63 Gensequenzen, von denen 59 verwertbar waren, erstellt haben. Die 59 verwertbaren Gensequenzen zeigen eine Reihe bekannter Mutationen auf dem Spike-Protein, darunter die alten Bekannten N501Y und E484K, die schon Beta (b.1.351 – der 351ste Abkömmling von b.1) aufgewiesen hat und D614G, die erst-Mutation nach dem CCP-Urvirus, die alle Lineages enthalten. Dass die Gensequenzen nicht alle identisch sind, die Zuordnung zu C.1.2 auf einem eng umrissenen Set von Mutationen basiert, das wird anhand der folgenden Ergebnisse deutlich. Die Mutationen, die C.1.2 auszeichnen, finden sich vor allem in den Open Reading Frames (ORF) 1a, 3a und 9a, sowie in den Proteinen S(pike), E, M und N.
30 Mutationen, die C.1.2 bestimmen, finden bei mehr als der Hälfte der 57 Gensequenzen;
Auf dem Spike-Protein haben etwas mehr als 50% der zugeordneten Gensequenzen 14 Mutationen;
Rund 44% der 57 Gen-Sequenzen weisen die P25L-Mutation in der N-Terminal Domain (NTD) auf;
19% der 57 Gen-Sequenzen haben L585F (von Lambda bekannt) im Spike Protein;
16% haben die Mutation T478K in der S1-Domain des Spike-Proteins (von Delta bekannt);
8% tragen die Mutation D936H und
weitere 8% die Mutation H1101Q in der S2-Domain des Spike-Proteins;
Eine Mehrheit der Gensequenzen, über die genaue Anzahl machen die Autoren keine Angabe, trägt nicht synonyme Mutationen in der N-terminal Domain (NTD), darunter Y144del, 242-244del, also Löschungen, die aus Alpha und Beta bekannt sind.
Insgesamt zählen die Autoren 57 für sie relevante Mutationen (unter gut 30.000 möglichen) von denen einige (Beispiele oben) bereits von anderen Varianten bekannt sind. Daraus, dass C.1.2 Mutationen aufweist, die sich auch für Alpha und Beta finden, schließen die Autoren, dass C.1.2 die Fähigkeit mitbringen KÖNNTE, Impfstoffen zu entgehen und daraus, dass C.1.2 weitere Mutationen auf dem Spike-Protein mit Eta und Gamma teilt, schließen sie, dass die Gefahr, dass Impfstoffe nicht wirksam sind, sehr hoch ist. Daraus, dass die Verbreitung von C.1.2 in Südafrika, wie sie schreiben: konsistent steigt, von einem Anteil von 0,2% unter den im Mai in Südafrika gesammelten Gen-Sequenzen, zu einem Anteil von 1,6% im Juni und 2,0% im Juli, folgern die Autoren, eine höhere Mutationsgeschwindigkeit und aus all dem leiten sie ab, dass C.1.2 nicht, wie das Public Health England schreibt, eine Variant of Investigation, sondern eine Variant of Concern sei.
Und der mediale Widerhall ist erstaunlich, geradezu orchestriert. Es wäre eine eigene Forschung wert, herauszufinden, wie es ein Beitrag, der am 26. August auf medRxiv veröffentlich wurde (und damit nicht peer reviewd ist – was für Redations-Schranzen ja gemeinhin sehr wichtig ist.), in so kurzer Zeit in Presseagenturen und danach in fast alle Zeitungen Deutschlands schafft. Man muss den Eindruck gewinnen, dass es bei Agenturen Beschäftigte gibt, die regelrecht darauf lauern, dass eine neue Studie, die verspricht, eine noch gefährlichere Variante zu Tage befördert zu haben, auftaucht, die man dann in aller Unkenntnis und mit nur einem Ziel: Panik, breittreten kann.
Indes sind die Ergebnisse von Scheepers et al. (2021) bestenfalls vorläufig, denn die Zuordnung ihrer Gensequenzen zu C.1.2 ist eine mittels Pangolin, also über eine Software, die nach Gemeinsamkeiten such, und zwar auf Grundlage von Gensequenzen, die die Anforderungen an eine eindeutige Bestimmung nicht erfüllen, weil sie weniger als 95% des Genoms von SARS-CoV-2 umfassen. Für diese unvollständigen Gensequenzen, so haben O’Toole et al. (2021) in ihrem Beitrag gezeigt, die häufig nur aus dem Spike-Protein bestehen, ist es wichtig, einem bestimmten von O’Toole et al. (2021) entwickelten Prozedere zu folgen, um sichergehen zu können, dass die Zuschreibung zu einer Pango Lineage auch korrekt ist. O’Toole haben ihren Beitrag am 10. August 2021 veröffentlicht. Der Beitrag findet sich nicht in der Literaturliste von Scheepers et al. (2021). Sie sind der Verfahrensweise, die O’Toole et al. (2021) entwickelt haben, also nicht gefolgt. Der Stellenwert der Ergebnisse von Scheepers et al. (2021) ist damit unklar, vermutlich dubios.
Für Dilettanten in Medien und Politik, die es gewohnt sind, sich über Dinge auszulassen, von denen sie keine Ahnung haben, ist das offenkundig kein Problem.
Übrigens haben Scheepers et al. (2021) keinerlei Antwort auf die Frage gegeben, ob C.1.2 gefährlicher als bisherige Varianten ist. Sie interessieren sich ausschließlich für das vielleicht vorhandene Potential, Impfstoffen zu entkommen. Jeder Schreiberling, der etwas anderes behauptet, hat seiner Phantasie freien Lauf gelassen.
Scheepers, Cathrine et al. (2021). The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection. medRxiv.
Folgen Sie uns auf Telegram.
Anregungen, Hinweise, Kontakt? -> Redaktion @ Sciencefiles.org
Wenn Ihnen gefällt, was Sie bei uns lesen, dann bitten wir Sie, uns zu unterstützen.
ScienceFiles lebt weitgehend von Spenden.
Helfen Sie uns, ScienceFiles auf eine solide finanzielle Basis zu stellen.Wir haben drei sichere Spendenmöglichkeiten:
Mein spontaner Gedanke beim Lesen dieses Beitrags zur allgemeinen Diskussion:
Die genannte Software heißt ‘Pangoline’ … und gerade zu Anfang des P(l)andemie-Theaters wurde regelmäßig das Schuppentier (Pangoline) als Virus-Ursprung diskutiert.
Frei nach Dr. Diefenbach -ob aus Dummheit oder Böswilligkeit sei dahin gestellt- was wäre wenn, schlicht bewusst oder unbewusst im Zuge von Übersetzungsfehlern / Medienrummel der kognitiven Minderleister aus der Software ‘Pangoline’ das Schuppentier ‘Pangoline’ wurde???
… oder sollte diese Wortgleichheit tatsächlich nur ein reiner Zufall unter tausenden möglichen Tierarten sein?
So ne kleine Buchstaben-Mutation ist doch unerheblich. Das Virchen stammt halt doch aus dem (Computer)-Labor. Passt wunderbar zu der ganzen Fake-P(l)andemie.
High, wenn man ein Feuer neu anfachen will, braucht man einen neuen Holz-Scheit und etwas Wind. Wenn man eine Panik neu anfachen will, braucht man wohl heute nur eine neue Virus-Variante, die theoretisch nur eine neue Bezeichnung haben muss. Dann genügt es wohl, viel Wind darum zu machen. Der Gewinn ist dann heiße Luft. 🙂 Freundlichst Fiete
“der mediale Widerhall ist erstaunlich, geradezu orchestriert”
Angesichts der Tatsache, daß die meisten Medien die vorformulierten Meinungen der sogenannten Nachrichten Agenturen wörtlich übernehmen, kein wirkliches Wunder. Die Gleichschaltung ist längst Wirklichkeit!
Was dann auch sehr deutlich macht, daß es bei der sogenannten Pandemie spätestens nach den ersten zwei Monaten eigentlich nur um eine Diffamierung des Gesunden und des Individuums an sich geht!
Ja, Herr Feldmann, Willkommen in der schönen neuen Welt, wo sich gerade ein Teil des “homo sapiens sapiens” in die Dekadenz und Degeneration verabschiedet.
Ich habe inzwischen als positive ERwartungshaltung, daß die Evolution ihren Job macht.
Die biologischen “Mechanismen” schlucken manches weg und sind allgemein sehr flexibel, aber einiges funktioniert kategorisch nicht: Königswasser trinken etc. Und ebenso dürfte sich mit jeglicher Gentherapie so sie nicht gründlichst geprüft und entwickelt wird (das gilt für den ganzen Corona-Zirkus in keiner Weise) eine Büchse der Pandora öffnen, die evtl. wirklich zum Zusammenbruch des Immunsystems der mit diesem Müll aufgefüllten Menschen führt. Die Welt leidet keinen Verlust, wenn die Spahns,Merkels,Lauterbachs,Söders etc.pp, es sind viele, von uns gehen! Für mich sind die schon tot. Die, die die Aufgabe der Humanität vorantreiben*, werden durch ihr Verwehen zur Renaissance des Humanen.
*Ich meine nicht nur die Diffamierung der Freiheit und des Gesunden (interessant wenn man Nietzsches Anmerkungen in diese Zeit hält!), sondern explizit solches Fäkaldenken, daß man “Ungeimpfte nicht mehr medizinisch behandeln soll” oder “Ungeimpfte einsperren”, “PCR-Positive nicht mehr reanimieren” usf. .
Wer sowas äußert, ist für eine zivile Gesellschaft verbrannt.
Mit anderen Worten: “Wir haben überhaupt keine Verwendung für Sie, da wir Menschen sind, was Sie zu sein abgelehnt haben. Hauen Sie ab ! Verschwinden Sie ! Gehen Sie und verrecken Sie ! Sonst werden wir alle sterben wegen Ihrer idiotischen Anmaßung und Ihrer Entfremdung von einer Wirklichkeit, die Sie uns durch Ihr Pflichtversäumnis aufgebürdet haben !” Zitatende (Philip Wylie: “Das Wundertier”).
Ja, ungefähr so, sehr geehrte Erinnerung! Ich bin bloß literarischer Selbstdreher und muß mit meinen kargen Worten vorlieb nehmen, ohne so ein Munitionslager wie Sie im Hintergrund zu haben.
In Anotation zu Ihrem Nickname müßte ich für mich den “Vergessene Wahrheit” wählen.
Dafür treffe ich dann gelegentlich auf kulturelle Erzeugnisse, die meine Misanthropie in ihren Grundfesten erschüttern, wie bspw. die gentle of my mind Interpretations der jüngsten The Petersens Schwester dieser Tage
Sehr geehrter Herr Feldmann, das “Munitionslager” ist ein Erbstück meines Vaters, das von mir noch einmal aufgestockt wurde. Glücklicherweise hat mich die assoziative “Erinnerung” an diese “Vergessenen Wahrheiten” nicht verlassen, so daß automatisch “nachgeladen” wird.
Worte sind für mich Phoneme, die mir andeuten, durch welchen Filter jemand diese Konsensrealität darstellen will: Sofern derjenige tatsächlich meint, was er sagt und ich verstehe, was gemeint sein soll (“Never saying what they mean – never meaning what they say”- Games people play, Liedtext: Joe South).
Solange ich nicht weiß, was ich will, wissen es andere für mich; und das, was sie von sich geben, ist von ihnen selbst häufig unverdaut (‘Büttel’). So grüßt eben täglich das Murmeltier. Diesen Reduktionismus teile ich nicht.
Worte sind für mich kein Problem, lediglich die Erwartungen sogenannter “Erwachsener mündiger Bürger”, daß und wie ich auf ihre Worte reagiere: Etwa auf ihren “freiwilligen” Zwang, mir Injektionen unterjubeln zu lassen.
Nun liegt mir Pazifismus fern. Pazifisten vertreten die Auffassung, der Wolf sei Vegetarier. Deshalb habe ich “nicht verweigert” und bin gerne “in die Wolken” gegangen. Dort kommt man eventuell auf Gedanken wie Alexander Gerst, Richard Bach (Möve Jonathan) oder Saint-Exupéry.
Eventuell, denn ich habe auch miterlebt, daß sich (1970) die Passagiere bei bester Sicht aus 11 km Höhe in einer 747 lieber einen Film mit Romy Schneider ‘reinzogen – wobei sie die Sichtblenden nach draußen schlossen um nicht geblendet zu werden. Das war Platos Höhlengleichnis “life”.
Ohne den Kontakt zu diesen Menschen zu verlieren, bin ich zum Autodidakten geworden und freue mich über die Eindrücke, mit denen dieser beeindruckende Kosmos aufwartet (Kant:”Der bestirnte Himmel über mir und das moralische Gesetz in mir”), sofern man sich nicht von aufgeblasenen Tölpeln die Sicht darauf versperren läßt.
Ich kann zwar manchmal gehörig betroffen sein, wenn Sprüche kommen wie: “Ich habe dich immer beneidet und gehaßt wegen deines inneren Friedens und deiner Lebensfreude und das wollte ich dir kaputtmachen”. Das sind die lebenden Toten, die man ihre Toten begraben lassen soll. Die reichen aber nicht aus, um zum Misanthrop zu werden, nur zum Menschendurchschauer, wie beispielsweise Schopenhauer und Nietzsche es waren. Das kann dann tatsächlich zu einer persönlichen ‘Renaissance’ führen.
Le deseo mucha salud y mucha fuerza y que viva muchos años mas para que mas niños puedan encontrarlo en la sala de espera.
Es gab Generationen, die echte Gefahren erlebten: Bombennächte, Artilleriefeuer, Flucht, marodierende Soldaten, Seuchen usw. usw. Herangewachsen ist inzwischen eine Generation, die im Fernsehen oder in Videospielen danach lechzt, Gewalttaten und Schrecken zu erleben. Es ist gewiss kein Zufall, dass in der Nachkriegszeit Heimatfilme Konjunktur hatten und heute gemetzelt wird was der Bildschirm hergibt. Es muss wunderbar sein, sich in der Realität, wenn auch nur in der vorgestellten Realität, einer fürchterlichen Gefahr zu stellen, dem Piks in der Oberarm mutig ins Auge zu blicken und hinterher davon zu berichten. Bin schon gespannt, wie diese Farce weiter geht. In Israel wird ja inzwischen zum dritten Schuss aufgerufen, weil die ersten beiden verpufft sind, und es wird gedroht, dass die doppelt Geimpften bald so behandelt werden könnten wie die Ungeimpften, wenn nicht alle schnellstens zum Schuss antreten. Warum nicht wird nicht gleich ein Abonnement angeboten mit kostenlosem Toaster für schnell Entschlossene?
Ich kann mir gut vorstellen, dass diese Variante schon sehnsüchtig erwartet wird. Die wäre dann der Deus ex Machina für die Politik, wenn die Inzidenzen unter den Geimpften wieder ansteigen.
Ich glaube eher, dass sie sehnsüchtig kreiert wurde! Denn: Man will jetzt noch mehr Schwung in den Impfkrieg bringen: Der Krankheitsminister Jens Spahn-Wahn und NRW-Arbeitgeberpräsident Arndt Kirchhoff wollen nun eine 2G-Regelung auch für Betriebe einführen. “Klar sei, dass die Impfverweigerung Einzelner nicht zum Schaden von Kollegen, Arbeitgebern und Kunden sein dürfe – auch wenn Unternehmen rechtlich niemanden zum Impfen zwingen könnten.” Zynismus Ende
Klar ist nur, dass wir Ungeimpften krank sind… Ja, impfen ist so geil – und die Hirne der Covid19-Religions-Fanatiker auf Erbsengröße geschrumpft…
Hell is empty and the devils are all here. William Shakespeare
Das kommt mir irgendwie vor wie die Waschmittelwerbung in den 1970ern. Da hat auch jedes Mittel noch weißer noch weißer gewaschen als das Vorige. Blöd nur, wenn die Textilien vorher gar nicht als weiß gedacht waren.
Ehrlicherweise muss ich aber zugeben, dass der Begriff “Delta” schon Eindruck machte, indem er einfach Assoziationen zur effizienten Tötungsmaschinerie “Delta Force” wecken konnte.
Da wird einem das Virus schon suspekt – genau wie Hurricanes die weibliche Namen haben mehr Schäden und Tote verursachen. Nicht, dass die Winde mit Frauennamen schlimmer seien, nein die Leute nehmen diese Hurricanes dann weniger ernst. Scheint ein menschliches Phänomen zu sein.
C 1.2 klingt aber eher wie ein Flugzeug oder ein Zug in einem Schachspiel.
Da hätten die lieber einen anderen Namen nehmen sollen. Z.B.: Hiobsvariante oder Nemesisvariante oder so….
Der Propaganda-Maschinerie gehen das Geld und die Ideen aus wie es ausschaut. Vielleicht haben die sich zu viel von ihren eigenen Drogen reingespritzt und die Hirnvenenthrombose wirkt bei hirnlosen natürlich nicht tödlich sondern führt zu solchen brainfarts.
“…wenn sie die 345.356 SARS-CoV-2 Genome, die in GISAID gespeichert sind, analysieren. Unter 458.622 Gensequenzen von SARS-CoV-2, die einer Lineage im Pango System zugeordnet sind, finden die Autoren 31.940 Nukleotid-Sequenzen, die dem Spike-Protein zugeordnet sind…”
In der Virologie bin ich ein absoluter Laie. Aber vielleicht bin ich nicht der einzige, der diese Zahlen nicht versteht.
Was heißt: 345.356 SARS-CoV-2 Genome? Ich denke, ein Virus hat ein Genom. Oder gibt es mittlerweile so viele Varianten?
Was heißt: 458.622 Gensequenzen von SARS-CoV-2? Ich denke, das Genom hat 30.000 Basenpaare, und je nachdem kann ich diese in Sequenzen von verschiedenen Längen aufteilen.
Lieber József, die Virologen verstehen es auch nicht. Lese lieber meinen folgenden Beitrag. Das ist verständlich und ein ungefährliches Geschäftsmodell ungeahnten Ausmaßes.
Ich bin Hobby-Virologe und habe etwas ganz neues, aber dafür ungefährliches Virus entdeckt, was für den Nobel-Pteis garantiert reicht. Ungeimpfte infizieren ungeimpfte und bekommen dafür den grünen Pass, als Genesene. UGI.1.2.3 ist die Zauberformel und nicht aus der Corona-Familie. Wenn ich mich irren sollte, wäre es aber eine Super-Geschäftsidee. Muss demnächst Onkel Bill anrufem.
Wir sehen, dass du dich in Vereinigtes Königreich befindest. Wir haben unsere Preise entsprechend auf Pfund Sterling aktualisiert, um dir ein besseres Einkaufserlebnis zu bieten. Stattdessen Euro verwenden.Ausblenden
 Die MS-Medien-Euphorie ist ausgebrochen:
Die MS-Medien-Euphorie ist ausgebrochen: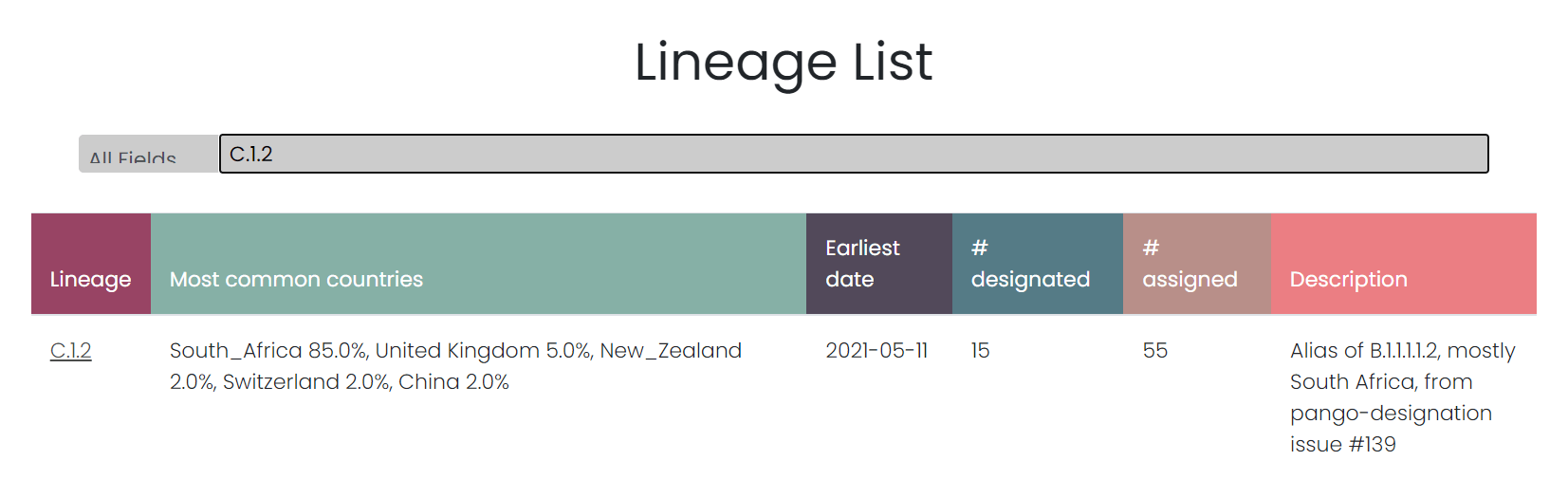
 Der Fundus von C.1.2 besteht derzeit aus 70! Sequenzen, davon sind 15 Sequenzen als C.1.2 ausgewiesen, 55 sind C.1.2 nur zugeordnet. Ein wichtiger Unterschied, zu dem wir noch kommen. C.1.2 ist eine Bezeichnung, die der Nomenklatura der sogenannte Pango Lineage entstammt, einem Versuch, das Mutationschaos, das sich bei RNA-Viren und DNA-Viren einstellt, mehr oder minder in den Griff zu bekommen. Die Zuordnung einer Sequenz zu einer Lineage ist klar geregelt. Es gibt ein Set von Regeln, das über 10 Seiten ausgebreitet wird, und das hier nachgelesen werden kann. Notwendig ist diese Nomenklatur, weil das Genom von SARS-CoV-2 rund 30.000 Nukleotide umfasst und somit rund 30.000 potentielle Orte, an denen sich eine Mutation einstellen kann. Nicht nur können sich an 30.000 verschiedenen Stellen im Genom von SARS-CoV-2 Mutationen einstellen, es können sich auch in unterschiedlichen Sequenzen an unterschiedlichen Stellen unterschiedlich viele Mutationen einstellen. Deshalb ist es notwendig, eine Methode zu finden, um die vielen möglichen Mutationen, die sich auf einem Genom oder auf einer Genomsequenz einfinden können, zuzuordnen oder genau auszuweisen.
Der Fundus von C.1.2 besteht derzeit aus 70! Sequenzen, davon sind 15 Sequenzen als C.1.2 ausgewiesen, 55 sind C.1.2 nur zugeordnet. Ein wichtiger Unterschied, zu dem wir noch kommen. C.1.2 ist eine Bezeichnung, die der Nomenklatura der sogenannte Pango Lineage entstammt, einem Versuch, das Mutationschaos, das sich bei RNA-Viren und DNA-Viren einstellt, mehr oder minder in den Griff zu bekommen. Die Zuordnung einer Sequenz zu einer Lineage ist klar geregelt. Es gibt ein Set von Regeln, das über 10 Seiten ausgebreitet wird, und das hier nachgelesen werden kann. Notwendig ist diese Nomenklatur, weil das Genom von SARS-CoV-2 rund 30.000 Nukleotide umfasst und somit rund 30.000 potentielle Orte, an denen sich eine Mutation einstellen kann. Nicht nur können sich an 30.000 verschiedenen Stellen im Genom von SARS-CoV-2 Mutationen einstellen, es können sich auch in unterschiedlichen Sequenzen an unterschiedlichen Stellen unterschiedlich viele Mutationen einstellen. Deshalb ist es notwendig, eine Methode zu finden, um die vielen möglichen Mutationen, die sich auf einem Genom oder auf einer Genomsequenz einfinden können, zuzuordnen oder genau auszuweisen.

 Für diese neue Lineage gibt es derzeit 70 Sequenzen. Das hindert die Hirnis, die in Redaktionen sitzen, aber nicht daran, schon einmal den Panikknopf umzustellen und ihr übliches Lamento anzustimmen, das, das sie immer anstimmen: gefährlicher, übertragbarer, tödlicher. Alle Beiträge haben wiederum die gerade von Catherine Scheepers und … bis Jinal N. Bhiman (2021) veröffentlichte Arbeit mit dem Titel “The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection” zum Gegenstand, in der die Autoren von ihren Analysen berichten, die sie auf Basis von 63 Gensequenzen, von denen 59 verwertbar waren, erstellt haben. Die 59 verwertbaren Gensequenzen zeigen eine Reihe bekannter Mutationen auf dem Spike-Protein, darunter die alten Bekannten N501Y und E484K, die schon Beta (b.1.351 – der 351ste Abkömmling von b.1) aufgewiesen hat und D614G, die erst-Mutation nach dem CCP-Urvirus, die alle Lineages enthalten. Dass die Gensequenzen nicht alle identisch sind, die Zuordnung zu C.1.2 auf einem eng umrissenen Set von Mutationen basiert, das wird anhand der folgenden Ergebnisse deutlich. Die Mutationen, die C.1.2 auszeichnen, finden sich vor allem in den Open Reading Frames (ORF) 1a, 3a und 9a, sowie in den Proteinen S(pike), E, M und N.
Für diese neue Lineage gibt es derzeit 70 Sequenzen. Das hindert die Hirnis, die in Redaktionen sitzen, aber nicht daran, schon einmal den Panikknopf umzustellen und ihr übliches Lamento anzustimmen, das, das sie immer anstimmen: gefährlicher, übertragbarer, tödlicher. Alle Beiträge haben wiederum die gerade von Catherine Scheepers und … bis Jinal N. Bhiman (2021) veröffentlichte Arbeit mit dem Titel “The continuous evolution of SARS-CoV-2 in South Africa: a new lineage with rapid accumulation of mutations of concern and global detection” zum Gegenstand, in der die Autoren von ihren Analysen berichten, die sie auf Basis von 63 Gensequenzen, von denen 59 verwertbar waren, erstellt haben. Die 59 verwertbaren Gensequenzen zeigen eine Reihe bekannter Mutationen auf dem Spike-Protein, darunter die alten Bekannten N501Y und E484K, die schon Beta (b.1.351 – der 351ste Abkömmling von b.1) aufgewiesen hat und D614G, die erst-Mutation nach dem CCP-Urvirus, die alle Lineages enthalten. Dass die Gensequenzen nicht alle identisch sind, die Zuordnung zu C.1.2 auf einem eng umrissenen Set von Mutationen basiert, das wird anhand der folgenden Ergebnisse deutlich. Die Mutationen, die C.1.2 auszeichnen, finden sich vor allem in den Open Reading Frames (ORF) 1a, 3a und 9a, sowie in den Proteinen S(pike), E, M und N.


 ScienceFiles Spendenkonto:
HALIFAX (Konto-Inhaber: Michael Klein):
ScienceFiles Spendenkonto:
HALIFAX (Konto-Inhaber: Michael Klein):


 ScienceFiles Spendenkonto:
HALIFAX (Konto-Inhaber: Michael Klein):
ScienceFiles Spendenkonto:
HALIFAX (Konto-Inhaber: Michael Klein):



Habe ich das richtig verstanden?
Radio Hamburg ist eine neue Corona Variante?
Wie wärs mit einer Impfung mit Zappelstrom von der Küste?
Bitte schnell anhören, bevors gelöscht wird:
Karl! Karl! Tu was!
Karl kann gerade nicht, der ist mit Jens und Saskia auf’m Klo. Kann ich irgendwas für Sie machen?
Mein spontaner Gedanke beim Lesen dieses Beitrags zur allgemeinen Diskussion:
Die genannte Software heißt ‘Pangoline’ … und gerade zu Anfang des P(l)andemie-Theaters wurde regelmäßig das Schuppentier (Pangoline) als Virus-Ursprung diskutiert.
Frei nach Dr. Diefenbach -ob aus Dummheit oder Böswilligkeit sei dahin gestellt- was wäre wenn, schlicht bewusst oder unbewusst im Zuge von Übersetzungsfehlern / Medienrummel der kognitiven Minderleister aus der Software ‘Pangoline’ das Schuppentier ‘Pangoline’ wurde???
… oder sollte diese Wortgleichheit tatsächlich nur ein reiner Zufall unter tausenden möglichen Tierarten sein?
Pangolin, nicht Pangoline
Phylogenetic Assignment of Named Global Outbreak Lineages
So ne kleine Buchstaben-Mutation ist doch unerheblich. Das Virchen stammt halt doch aus dem (Computer)-Labor. Passt wunderbar zu der ganzen Fake-P(l)andemie.
Oder das Virchen eben doch aus dem Computer-Pangolin stammt, sprich von Anfang an virtuell war, was ich sowieso schon lange vermute.
High, wenn man ein Feuer neu anfachen will, braucht man einen neuen Holz-Scheit und etwas Wind. Wenn man eine Panik neu anfachen will, braucht man wohl heute nur eine neue Virus-Variante, die theoretisch nur eine neue Bezeichnung haben muss. Dann genügt es wohl, viel Wind darum zu machen. Der Gewinn ist dann heiße Luft. 🙂 Freundlichst Fiete
Ein Abstrich verleiht Flügel! Er beflügelt die Fantasie!
Oder bringt die Nase zum Laufen. Was ja auch irgendwie beflügelt.
“der mediale Widerhall ist erstaunlich, geradezu orchestriert”
Angesichts der Tatsache, daß die meisten Medien die vorformulierten Meinungen der sogenannten Nachrichten Agenturen wörtlich übernehmen, kein wirkliches Wunder. Die Gleichschaltung ist längst Wirklichkeit!
Fakten:
https://www.naturalnews.com/2021-08-25-fda-gaslights-the-world-with-fake-approval-of-pfizer-vaccine.html
https://notrickszone.com/2021/08/26/vaccinated-27x-more-likely-to-be-symptomatically-infected-than-unvaccinated-whove-had-covid/
https://chriswaldburger.substack.com/p/bombshell-uk-data-destroys-entire
Kontra Irrsinn:
https://theconservativetreehouse.com/blog/2021/08/25/the-science-of-hope-biden-administration-will-promote-new-plan-for-multiple-booster-shots-every-six-months-in-perpetuity-for-covid-variants/#more-215887
DAS HEIẞT: ALS GESUNDER IST MAN MEDIZINISCH AUS DEM SCHNEIDER –
P O L I T I S C H DAMIT LEIDER NICHT !
Was dann auch sehr deutlich macht, daß es bei der sogenannten Pandemie spätestens nach den ersten zwei Monaten eigentlich nur um eine Diffamierung des Gesunden und des Individuums an sich geht!
Medizinisch ist da eher nichts bis wenig.
Ja, Herr Feldmann, Willkommen in der schönen neuen Welt, wo sich gerade ein Teil des “homo sapiens sapiens” in die Dekadenz und Degeneration verabschiedet.
Ich habe inzwischen als positive ERwartungshaltung, daß die Evolution ihren Job macht.
Die biologischen “Mechanismen” schlucken manches weg und sind allgemein sehr flexibel, aber einiges funktioniert kategorisch nicht: Königswasser trinken etc. Und ebenso dürfte sich mit jeglicher Gentherapie so sie nicht gründlichst geprüft und entwickelt wird (das gilt für den ganzen Corona-Zirkus in keiner Weise) eine Büchse der Pandora öffnen, die evtl. wirklich zum Zusammenbruch des Immunsystems der mit diesem Müll aufgefüllten Menschen führt. Die Welt leidet keinen Verlust, wenn die Spahns,Merkels,Lauterbachs,Söders etc.pp, es sind viele, von uns gehen! Für mich sind die schon tot. Die, die die Aufgabe der Humanität vorantreiben*, werden durch ihr Verwehen zur Renaissance des Humanen.
*Ich meine nicht nur die Diffamierung der Freiheit und des Gesunden (interessant wenn man Nietzsches Anmerkungen in diese Zeit hält!), sondern explizit solches Fäkaldenken, daß man “Ungeimpfte nicht mehr medizinisch behandeln soll” oder “Ungeimpfte einsperren”, “PCR-Positive nicht mehr reanimieren” usf. .
Wer sowas äußert, ist für eine zivile Gesellschaft verbrannt.
Mit anderen Worten: “Wir haben überhaupt keine Verwendung für Sie, da wir Menschen sind, was Sie zu sein abgelehnt haben. Hauen Sie ab ! Verschwinden Sie ! Gehen Sie und verrecken Sie ! Sonst werden wir alle sterben wegen Ihrer idiotischen Anmaßung und Ihrer Entfremdung von einer Wirklichkeit, die Sie uns durch Ihr Pflichtversäumnis aufgebürdet haben !” Zitatende (Philip Wylie: “Das Wundertier”).
Ja, ungefähr so, sehr geehrte Erinnerung! Ich bin bloß literarischer Selbstdreher und muß mit meinen kargen Worten vorlieb nehmen, ohne so ein Munitionslager wie Sie im Hintergrund zu haben.
In Anotation zu Ihrem Nickname müßte ich für mich den “Vergessene Wahrheit” wählen.
Dafür treffe ich dann gelegentlich auf kulturelle Erzeugnisse, die meine Misanthropie in ihren Grundfesten erschüttern, wie bspw. die gentle of my mind Interpretations der jüngsten The Petersens Schwester dieser Tage
MgG PF
Sehr geehrter Herr Feldmann, das “Munitionslager” ist ein Erbstück meines Vaters, das von mir noch einmal aufgestockt wurde. Glücklicherweise hat mich die assoziative “Erinnerung” an diese “Vergessenen Wahrheiten” nicht verlassen, so daß automatisch “nachgeladen” wird.
Worte sind für mich Phoneme, die mir andeuten, durch welchen Filter jemand diese Konsensrealität darstellen will: Sofern derjenige tatsächlich meint, was er sagt und ich verstehe, was gemeint sein soll (“Never saying what they mean – never meaning what they say”- Games people play, Liedtext: Joe South).
Solange ich nicht weiß, was ich will, wissen es andere für mich; und das, was sie von sich geben, ist von ihnen selbst häufig unverdaut (‘Büttel’). So grüßt eben täglich das Murmeltier. Diesen Reduktionismus teile ich nicht.
Worte sind für mich kein Problem, lediglich die Erwartungen sogenannter “Erwachsener mündiger Bürger”, daß und wie ich auf ihre Worte reagiere: Etwa auf ihren “freiwilligen” Zwang, mir Injektionen unterjubeln zu lassen.
Nun liegt mir Pazifismus fern. Pazifisten vertreten die Auffassung, der Wolf sei Vegetarier. Deshalb habe ich “nicht verweigert” und bin gerne “in die Wolken” gegangen. Dort kommt man eventuell auf Gedanken wie Alexander Gerst, Richard Bach (Möve Jonathan) oder Saint-Exupéry.
Eventuell, denn ich habe auch miterlebt, daß sich (1970) die Passagiere bei bester Sicht aus 11 km Höhe in einer 747 lieber einen Film mit Romy Schneider ‘reinzogen – wobei sie die Sichtblenden nach draußen schlossen um nicht geblendet zu werden. Das war Platos Höhlengleichnis “life”.
Ohne den Kontakt zu diesen Menschen zu verlieren, bin ich zum Autodidakten geworden und freue mich über die Eindrücke, mit denen dieser beeindruckende Kosmos aufwartet (Kant:”Der bestirnte Himmel über mir und das moralische Gesetz in mir”), sofern man sich nicht von aufgeblasenen Tölpeln die Sicht darauf versperren läßt.
Ich kann zwar manchmal gehörig betroffen sein, wenn Sprüche kommen wie: “Ich habe dich immer beneidet und gehaßt wegen deines inneren Friedens und deiner Lebensfreude und das wollte ich dir kaputtmachen”. Das sind die lebenden Toten, die man ihre Toten begraben lassen soll. Die reichen aber nicht aus, um zum Misanthrop zu werden, nur zum Menschendurchschauer, wie beispielsweise Schopenhauer und Nietzsche es waren. Das kann dann tatsächlich zu einer persönlichen ‘Renaissance’ führen.
Le deseo mucha salud y mucha fuerza y que viva muchos años mas para que mas niños puedan encontrarlo en la sala de espera.
Fußnote:
https://brennstoff.com/artikel/der-bestirnte-himmel-ueber-mir-und-das-moralische-gesetz-in-mir-immanuel-kant/
Es gab Generationen, die echte Gefahren erlebten: Bombennächte, Artilleriefeuer, Flucht, marodierende Soldaten, Seuchen usw. usw. Herangewachsen ist inzwischen eine Generation, die im Fernsehen oder in Videospielen danach lechzt, Gewalttaten und Schrecken zu erleben. Es ist gewiss kein Zufall, dass in der Nachkriegszeit Heimatfilme Konjunktur hatten und heute gemetzelt wird was der Bildschirm hergibt. Es muss wunderbar sein, sich in der Realität, wenn auch nur in der vorgestellten Realität, einer fürchterlichen Gefahr zu stellen, dem Piks in der Oberarm mutig ins Auge zu blicken und hinterher davon zu berichten. Bin schon gespannt, wie diese Farce weiter geht. In Israel wird ja inzwischen zum dritten Schuss aufgerufen, weil die ersten beiden verpufft sind, und es wird gedroht, dass die doppelt Geimpften bald so behandelt werden könnten wie die Ungeimpften, wenn nicht alle schnellstens zum Schuss antreten. Warum nicht wird nicht gleich ein Abonnement angeboten mit kostenlosem Toaster für schnell Entschlossene?
Wobei es ja in Israel sogar noch den Hauch der realen Gefahr gibt.
Ich kann mir gut vorstellen, dass diese Variante schon sehnsüchtig erwartet wird. Die wäre dann der Deus ex Machina für die Politik, wenn die Inzidenzen unter den Geimpften wieder ansteigen.
Ich glaube eher, dass sie sehnsüchtig kreiert wurde! Denn: Man will jetzt noch mehr Schwung in den Impfkrieg bringen: Der Krankheitsminister Jens Spahn-Wahn und NRW-Arbeitgeberpräsident Arndt Kirchhoff wollen nun eine 2G-Regelung auch für Betriebe einführen. “Klar sei, dass die Impfverweigerung Einzelner nicht zum Schaden von Kollegen, Arbeitgebern und Kunden sein dürfe – auch wenn Unternehmen rechtlich niemanden zum Impfen zwingen könnten.” Zynismus Ende
Klar ist nur, dass wir Ungeimpften krank sind… Ja, impfen ist so geil – und die Hirne der Covid19-Religions-Fanatiker auf Erbsengröße geschrumpft…
Hell is empty and the devils are all here. William Shakespeare
Das kommt mir irgendwie vor wie die Waschmittelwerbung in den 1970ern. Da hat auch jedes Mittel noch weißer noch weißer gewaschen als das Vorige. Blöd nur, wenn die Textilien vorher gar nicht als weiß gedacht waren.
Dürfen wir an dieser Mutation bitte endlich alle sterben, oder wie lange soll diese weltweite Völkerverarschung noch weitergehen?
Ungeimpft, maskenfrei, Immunsystem durch Chemotherapie angegriffen -ich sollte seit mindestens sieben Monaten tot sein.
Statt dessen höre ich von der Wissenschaft nur könnte, eventuell, möglicherweise.
Früher begannen Märchen mit “Es war einmal”, heute mit “Die Wissenschaft sagt”.
Wie gut, daß Sie nicht daran geglaubt und d’ran geglaubt haben !!
Ehrlicherweise muss ich aber zugeben, dass der Begriff “Delta” schon Eindruck machte, indem er einfach Assoziationen zur effizienten Tötungsmaschinerie “Delta Force” wecken konnte.
Da wird einem das Virus schon suspekt – genau wie Hurricanes die weibliche Namen haben mehr Schäden und Tote verursachen. Nicht, dass die Winde mit Frauennamen schlimmer seien, nein die Leute nehmen diese Hurricanes dann weniger ernst. Scheint ein menschliches Phänomen zu sein.
C 1.2 klingt aber eher wie ein Flugzeug oder ein Zug in einem Schachspiel.
Da hätten die lieber einen anderen Namen nehmen sollen. Z.B.: Hiobsvariante oder Nemesisvariante oder so….
Der Propaganda-Maschinerie gehen das Geld und die Ideen aus wie es ausschaut. Vielleicht haben die sich zu viel von ihren eigenen Drogen reingespritzt und die Hirnvenenthrombose wirkt bei hirnlosen natürlich nicht tödlich sondern führt zu solchen brainfarts.
“…wenn sie die 345.356 SARS-CoV-2 Genome, die in GISAID gespeichert sind, analysieren. Unter 458.622 Gensequenzen von SARS-CoV-2, die einer Lineage im Pango System zugeordnet sind, finden die Autoren 31.940 Nukleotid-Sequenzen, die dem Spike-Protein zugeordnet sind…”
In der Virologie bin ich ein absoluter Laie. Aber vielleicht bin ich nicht der einzige, der diese Zahlen nicht versteht.
Was heißt: 345.356 SARS-CoV-2 Genome? Ich denke, ein Virus hat ein Genom. Oder gibt es mittlerweile so viele Varianten?
Was heißt: 458.622 Gensequenzen von SARS-CoV-2? Ich denke, das Genom hat 30.000 Basenpaare, und je nachdem kann ich diese in Sequenzen von verschiedenen Längen aufteilen.
Lieber József, die Virologen verstehen es auch nicht. Lese lieber meinen folgenden Beitrag. Das ist verständlich und ein ungefährliches Geschäftsmodell ungeahnten Ausmaßes.
Ich bin Hobby-Virologe und habe etwas ganz neues, aber dafür ungefährliches Virus entdeckt, was für den Nobel-Pteis garantiert reicht. Ungeimpfte infizieren ungeimpfte und bekommen dafür den grünen Pass, als Genesene. UGI.1.2.3 ist die Zauberformel und nicht aus der Corona-Familie. Wenn ich mich irren sollte, wäre es aber eine Super-Geschäftsidee. Muss demnächst Onkel Bill anrufem.